Improved knockout with Cas9
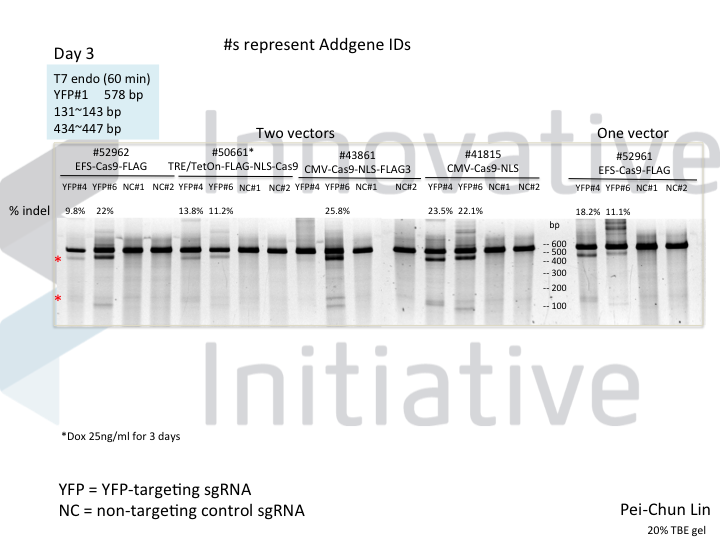
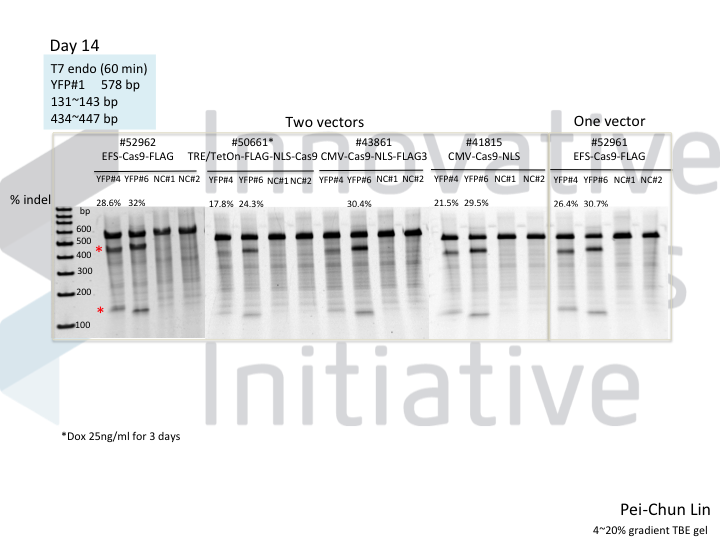
Cas9 is usually pretty good at gene knockout. Except when it isn’t. Most people who have gotten their feet wet with gene editing have had an experience like that in the following gel, in which so...
Cas9 is usually pretty good at gene knockout. Except when it isn’t. Most people who have gotten their feet wet with gene editing have had an experience like that in the following gel, in which some guides work very well but others are absolute dogs.
That’s a problem if you have targeting restrictions (e.g. when going after a functional domain instead of just making a randomly placed cut). So what can one do about it?
TL;DR Adding non-homologous single stranded DNA when using Cas9 RNP greatly boosts gene knockout.
The problem
There have been a few very nice papers showing that Cas9 prefers certain guides. I refer to these as the One True Guide hypothesis, with the idea being that Cas9 has somehow evolved to like some protospacers and dislike others. The data doesn’t lie, and there is indeed truth to this - Cas9 likes a G near the PAM and hates to use C. But guides that are highly active in one cell line are poor in others, and comparing very preference experiments in mouse cells vs worms gives very different answers. That’s not what you’d expect if the problem lies solely in Cas9’s ability to use a guide RNA to make a cut.
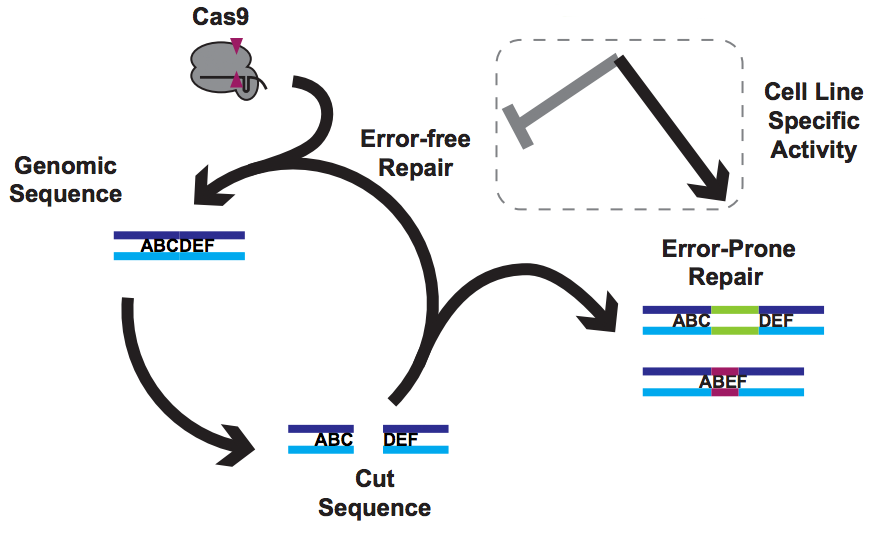
But of course, Cas9 is only making cuts. Everything else comes down to DNA repair by the host cell.
Our solution
In a new paper from my lab, just out in Nature Communications, we found that using a simple trick to mess with DNA repair can rescue totally inactive guides and make it easy to isolate knockout clones, even in challenging (e.g. polyploid) contexts. We call this approach “NOE”, for Non-homologous Oligonucleotide Enhancement.
(The acronym is actually a bit of a private joke for me, since I used to work with NOEs in a very different context, and Noe Valley is a nice little neighborhood in San Francisco)
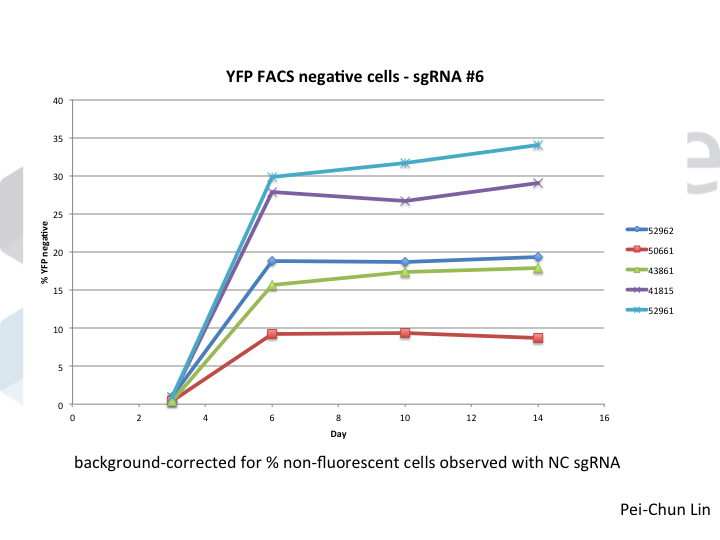
How does one perform NOE? It’s actually super simple. When using Cas9 RNPs for editing, just add non-homologous single stranded DNA to your electroporation reaction. That’s it. This increases indel frequencies several fold in a wide variety of cell lines and makes it easy to find homozygous knockouts even when using guides that normally perform poorly.
The key to NOE is having extra DNA ends. Single stranded DNA works the best, and even homologous ssDNAs one might use for HDR work. We tend to use ssDNAs that are not homologous to the human genome (e.g. a bit of sequence from BFP) because they make editing outcomes much simpler (NHEJ only instead of NHEJ + HDR). But double stranded DNAs also work, and even sheared salmon sperm DNA does the trick! Plasmids are no good, since there are no free ends.
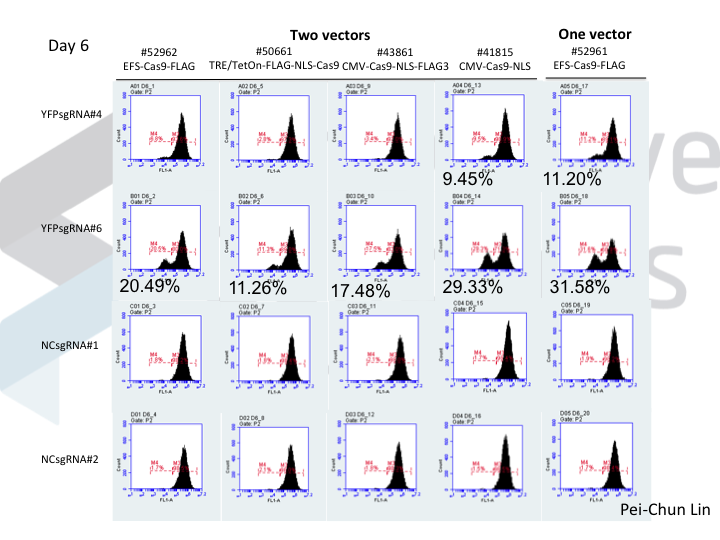
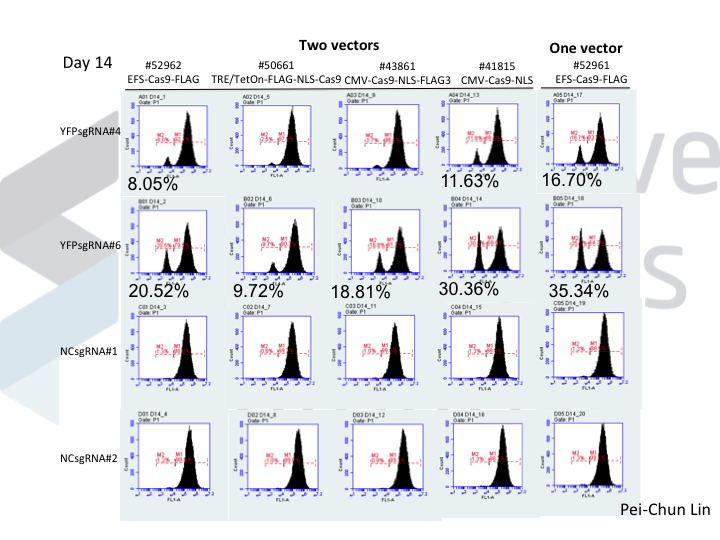
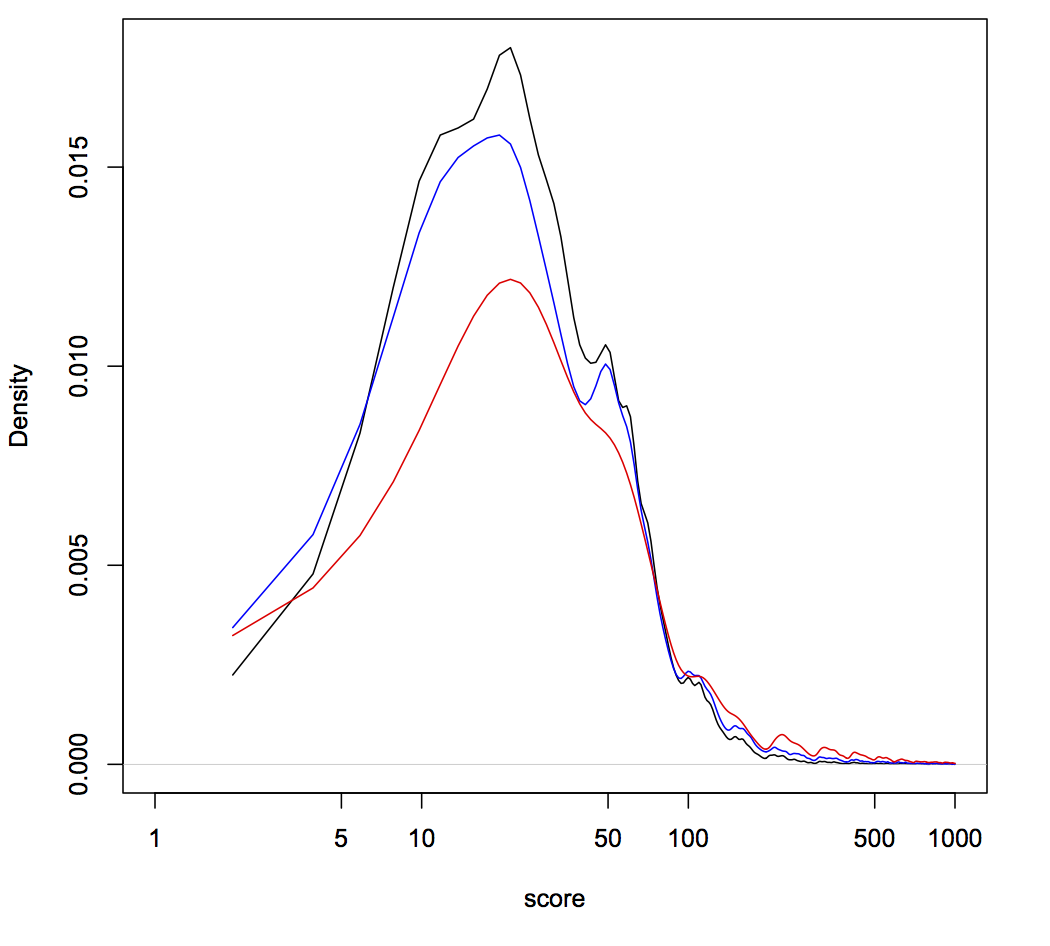
We know that NOE is doing something to DNA repair, because while this works in many cell lines, the molecular outcomes differ between cells! In many cells (5/7 that we’ve tested), NOE causes the appearance of very large deletions (much larger than you would normally see when using Cas9). But in 2/7 cells tested, NOE instead caused the cells to start scavenging little pieces of double stranded DNA and dropping them into the Cas9 break! The junctions of these pieces of DNA look like microhomologies, but we haven’t yet done the genetic experiments to say that this is caused by a process such as microhomology mediated end joining.
What's going on here?
How can making alterations in DNA repair so drastically impact the apparent efficacy of a given guide? We think that our data, together with data from other labs, implies that Cas9 cuts are frequently perfectly repaired. But this introduces a futile cycle, in which Cas9 re-binds and re-cuts that same site. The only way we observe editing is when this cycle is exited through imperfect repair, resulting in an indel. Perfect repair makes a lot of sense for normal DNA processing, since we accumulate DNA damage all the time in our normal lives. We'd be in a sorry state indeed if this damage frequently resulted in indels. It seems that NOE either inhibits perfect repair (e.g. titrating out Ku?) or enhances imperfect repair (e.g. stimulating an ATM response?), though we are still lacking direct data on mechanism at the moment.
What is it good for?
The ability to stimulate incorporation of double stranded DNA into a break might be useful, since non-homologous or microhomology-mediated integration of double stranded cassettes has recently been used for gene tagging. But we haven’t explicitly tried this. We have also found NOE to be very useful for arrayed screening, in which efficiency of the edit is key to phenotypic penetrance and subsequent hit calling.
Importantly, NOE seems to work in primary cells, including hematopoietic stem cells and T cells. We’ve been using it when doing pooled edits in unculturable primary human cells, and find that far higher fractions of cells have gene disruptions when using NOE. We’ve so far only worked in human cells with RNP, and I’m very interested to hear peoples’ experience using NOE in other organisms. We haven’t had much luck trying it with plasmid-based expression of Cas9, but other groups have told me that they can get it to work in that context as well.
How do I try it?
So if you’re interested, give it a shot. The details are all in our recent Nature Communications paper, but feel free to reach out if you have any more questions. This work was done by Chris Richardson (the postdoc who brought you flap-annealing HDR donors), Jordan Ray (an outstanding undergrad who is now a grad student at MIT), and Nick Bray (a postdoc bioinformatics guru).


 Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...
Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...