Why does fetal globin increase when editing beta globin? Published in Cell Reports
When my lab published editing of the sickle beta globin gene, together with Mark Walters and David Martin, we noticed something quite unexpected. Every time we targeted beta globin, we saw fetal globin go up! We also noticed this in beta globin editing papers from the labs of Paula Cannon and Matt Porteus. Transient stress can cause blood cells to express fetal globin, so at first we thought that we were just seeing something short-lived. But it turned out that fetal globin persisted after long-term transplantation of edited human cells into a mouse model. What could be happening in these cells? Could this be a route to treat globin-related diseases such as sickle cell disease? If so, why is beta thalassemia (lack of beta globin) not automatically compensated by fetal globin expression? A new paper from PhD student Mandy Boontanrart, out now in Cell Reports, finally figures out what’s going on. This work was completely driven by Mandy, using a wide range of techniques to dig into a difficult problem.
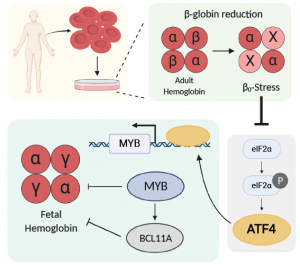
Editing itself can be stressful to cells, but Mandy’s first big findings were that the fetal globin response is not related to the the process of editing. Doing a scan of CRISPR cutting guides through beta globin, she found that any guide targeting an exon causes fetal globin to go up. But equally effective guides that target introns have no effect. She also found that CRISPRi transcriptional repression to knockdown beta globin does increase fetal globin. Therefore, it was something special about lacking beta globin, which is known as B0-stress.
Using clones of blood precursors edited to lack beta globin, Mandy did RNA-seq during differentiation to figure out how cells responded over time. The number one pathway by far was ATF4 signalling. But in a very strange way! It turned out that ATF4 signalling actually went down in cells with beta globin knockout. This was surprising because ATF4 responds to unfolded proteins, it had been suggested that lack of beta globin would lead to alpha chain aggregation that should turn on the unfolded protein response. We were seeing the exact opposite!
This actually makes sense in the context of recent work from the lab of Gerd Blobel, who found out that knockout of the heme-responsive kinase HRI can also increase fetal globin. HRI is upstream of eIF2a and ATF4, so it seemed that free heme (caused by lack of complete adult hemoglobin tetramers) had a stronger role than the free alpha chains in our system.
But what was ATF4 doing to increase fetal globin? Mandy investigated this using a large number of isogenically-controlled ChIP-seq experiments. She found that ATF4 doesn’t bind anywhere in the globin locus, nor does it bind anywhere near BCL11A, which is one of the latest hot globin regulators. In fact, Mandy found that ATF4’s effect on fetal globin still happened in K562 cells, which don’t express any BCL11A at all. Instead, Mandy found strong evidence that ATF4 regulates Myb through binding to a known enhancer region. Myb is itself a regulator of Bcl11A, but can also regulate fetal globin through other factors such as KLF1.
As always there’s a lot more to figure out. And it’s still unclear if we can use these findings to make a difference for patients with globinopathies. But thanks to Mandy’s work, we finally know why fetal globin increases when beta globin is reduced. We also have some clues as to why beta thalassemia is not always compensated by increased fetal globin. In fact, to some extent, it is! Many people with beta thalassemia exhibit increased fetal globin, but not enough to alleviate the symptoms of complete lack of beta globin. ATF4 does many jobs, so it could be that reducing ATF4 enough to yield large amounts of fetal globin might be bad for other biologies. This also adds a note of caution to targeting HRI as a therapeutic angle to increase fetal globin, since doing so could have pleiotropic effects. But all of that remains to be tested!
