INCREASING HEMOGLOBIN HBA2 BY REPAIRING THE HBD PROMOTER, PUBLISHED IN ELIFE
Erythrocytes, or red blood cells, carry hemoglobin and circulate throughout the body to supply oxygen. β-hemoglobinopathies, such as sickle cell disease...
Erythrocytes, or red blood cells, carry hemoglobin and circulate throughout the body to supply oxygen. β-hemoglobinopathies, such as sickle cell disease and β-thalassemia, are the most common genetic diseases worldwide and are caused by mutations affecting the structure or production of β-globin subunits in adult hemoglobin. These conditions result in anemia and organ damage, and available treatment options are limited. Stem cell transplantation is currently the only curative approach, although its feasibility relies on the availability of a suitable donor.
Hemoglobin is a tetrameric protein composed of 2 α-like (HBA) and 2 β-like subunits (HBB). Hemoglobin A1 (HbA1) constitutes 97% of adult hemoglobin, while Hemoglobin A2 (HbA2) makes up 2-3%. HbA2 is composed of two α-globin subunits and two δ-globin (HBD) subunits. HBD is a homologous to HBB gene, but with much lower expression compared to HBB due to a weak promoter. Currently, many efforts are focused on increasing fetal hemoglobin (HbF) to treat the β-hemoglobinopathies. But HbA2 is more similar to HbA1 and is already expressed at low levels in all adult red blood cells. What if we were to increase HbA2 levels? Could they potentially compensate for beta-globin deficiency? Can genome editing technologies be used to boost transcriptional activity of the endogenous HBD promoter to increase HbA2 levels? Mandy Boontanrart, a Postdoc in our lab, was eager to discover the answers to these questions.

Using CRISPR-Cas9 genome editing, we inserted various transcription factor binding sequences into the endogenous HBD promoter. Team efforts yielded positive results as we successfully increased the transcriptional activity in HUDEP-2 immortalized erythroid progenitor cells, resulting in a significant upregulation of HBD expression. Despite roughly equal homology-directed repair rates between all promoter designs, we observed a significant increase in HBD only for the design with all three elements (KLF1, β-DRF, and TFIIB). We next explored whether endogenous editing of the HBD promoter can be accomplished in bone marrow stem cells. We found up to 46% HBD expression in clonal populations. We also tested a small molecule drug that enhances HDR outcomes by inhibiting the NHEJ pathway and observed an increase in the percent of HDR alleles in pooled edited bone marrow stem cells.
While our findings provide key mechanistic insight into the globin gene regulation, several questions remain to be tackled. Is heterozygous knock-in of the promoter design in β-hemoglobinopathy cells is sufficient to ameliorate disease phenotypes? What is the safety profile of this strategy?
Overall, our work is a promising approach for restoring hemoglobin levels in red blood cells. This strategy might open new therapeutic avenues for to treating beta-hemoglobinopathies in the future.
For more, check out our paper, it is now out in Elife!
Note: Excitingly, Mandy is now leading an ETH spin-off, building upon the findings of the paper, check out their brand-new website https://www.ariyabio.ch/!
 ions causing the disorder. Homology directed repair (HDR)-based editing strategy as an option to correct FA mutations is pretty inefficient
ions causing the disorder. Homology directed repair (HDR)-based editing strategy as an option to correct FA mutations is pretty inefficient 
 Are you interested in
Are you interested in 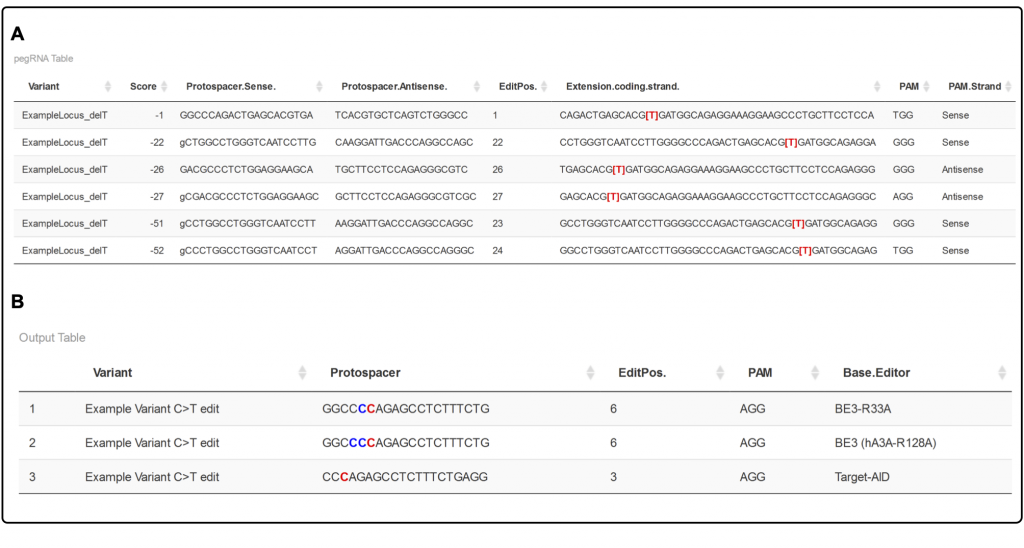
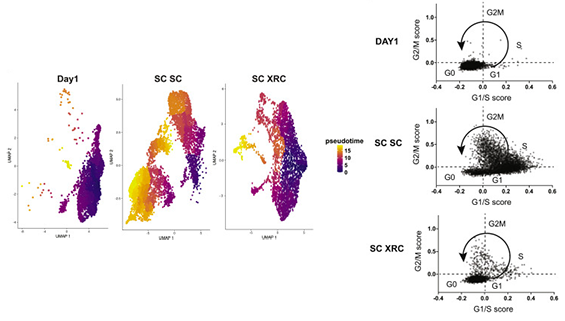 Jenny then had a very creative idea. She asked if a previously reported
Jenny then had a very creative idea. She asked if a previously reported 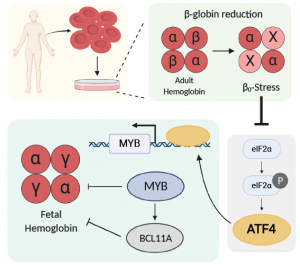
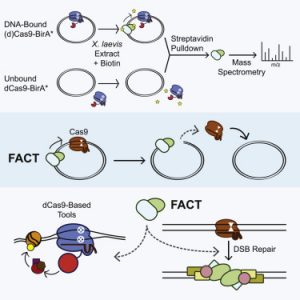 Alan took an unbiased approach to figure out what was removing Cas9 from DNA. He fused recombinant Cas9 with a promiscuous biotin ligase, bound purified Cas9-ligase to a plasmid, and used mass spectrometry to figure out what pushed the Cas9 off the plasmid. We were pleasantly surprised to find that both subunits of a dimeric histone chaperone called FACT were top mass spec hits! Follow-up experiments showed that FACT was necessary and sufficient for displacing Cas9 from DNA substrates. FACT was responsible for turning Cas9 from a multi-turnover “classic” nuclease enzyme into a single-turnover sticky enzyme!
Alan took an unbiased approach to figure out what was removing Cas9 from DNA. He fused recombinant Cas9 with a promiscuous biotin ligase, bound purified Cas9-ligase to a plasmid, and used mass spectrometry to figure out what pushed the Cas9 off the plasmid. We were pleasantly surprised to find that both subunits of a dimeric histone chaperone called FACT were top mass spec hits! Follow-up experiments showed that FACT was necessary and sufficient for displacing Cas9 from DNA substrates. FACT was responsible for turning Cas9 from a multi-turnover “classic” nuclease enzyme into a single-turnover sticky enzyme! The first set of hits are known homologous recombination factors, such as BRCA1. This gives high confidence that the screen worked as expected. Surprisingly, the same Fanconi Anemia complexes that are required for single stranded oligo HDR are required for plasmid HDR. The FA pathway is thus a core regulator of all forms of HDR!
The first set of hits are known homologous recombination factors, such as BRCA1. This gives high confidence that the screen worked as expected. Surprisingly, the same Fanconi Anemia complexes that are required for single stranded oligo HDR are required for plasmid HDR. The FA pathway is thus a core regulator of all forms of HDR! Our favorite CDC7 inhibitor is XL413, which is non-toxic and quite reversible. This distinguishes it from some other HDR-improving compounds that lead big genomic messes, including polyploidy. Delving into mechanism, CDC7 inhibition leads to loss of MCM2 phosphorylation. Because MCM2 phosphorylation is required for S phase progression, XL413 leads to a longer S phase. This is a magical phase of the cell cycle for HDR, and so our model is that XL413 increases HDR by increasing the amount of time cells are able to do HDR. We tested this with a timing experiment. Hitting cells with Cas9 and then immediately putting them in XL413 leads to increased HDR, because the cells are piling up in S phase at the same time they are repairing the Cas9 damage. But putting cells in XL413 first and then taking them out during editing leads to decreased HDR. This is because the cells all pile up S phase before editing, and then exit into HDR non-permissive cell cycle while Cas9 is doing its thing.
Our favorite CDC7 inhibitor is XL413, which is non-toxic and quite reversible. This distinguishes it from some other HDR-improving compounds that lead big genomic messes, including polyploidy. Delving into mechanism, CDC7 inhibition leads to loss of MCM2 phosphorylation. Because MCM2 phosphorylation is required for S phase progression, XL413 leads to a longer S phase. This is a magical phase of the cell cycle for HDR, and so our model is that XL413 increases HDR by increasing the amount of time cells are able to do HDR. We tested this with a timing experiment. Hitting cells with Cas9 and then immediately putting them in XL413 leads to increased HDR, because the cells are piling up in S phase at the same time they are repairing the Cas9 damage. But putting cells in XL413 first and then taking them out during editing leads to decreased HDR. This is because the cells all pile up S phase before editing, and then exit into HDR non-permissive cell cycle while Cas9 is doing its thing.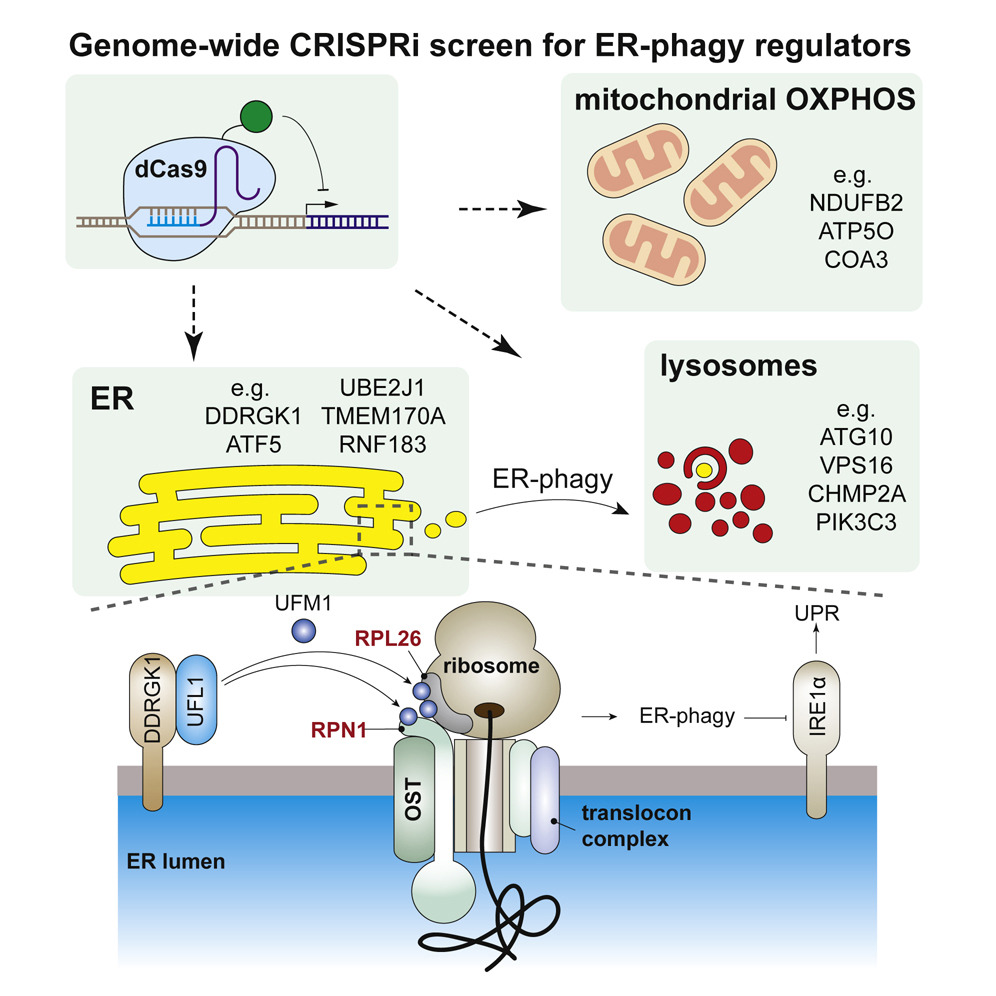 Did you know that cells eat their own organelles? This is best known when damaged mitochondria are degraded by autophagy (aka mitophagy). Failure to perform...
Did you know that cells eat their own organelles? This is best known when damaged mitochondria are degraded by autophagy (aka mitophagy). Failure to perform... Genome editing is all about DNA repair. So if you want to get your cells to do more of what you want (*cough* HDR *cough), you’d better know how they make ...
Genome editing is all about DNA repair. So if you want to get your cells to do more of what you want (*cough* HDR *cough), you’d better know how they make ...