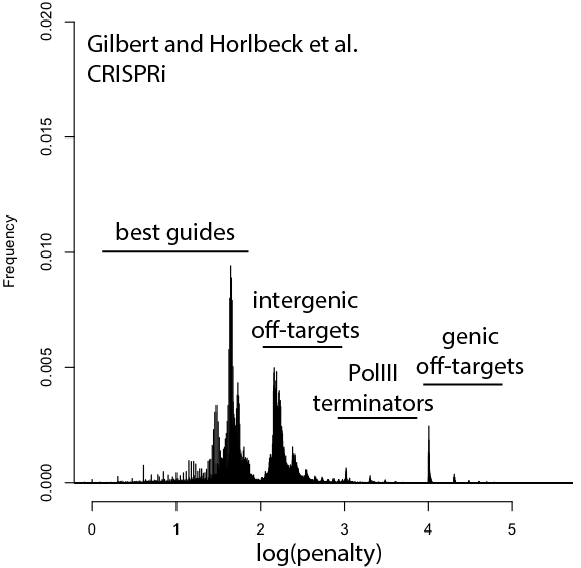
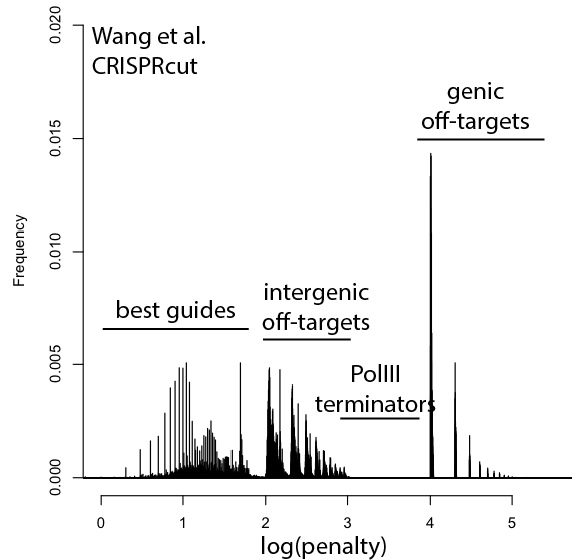
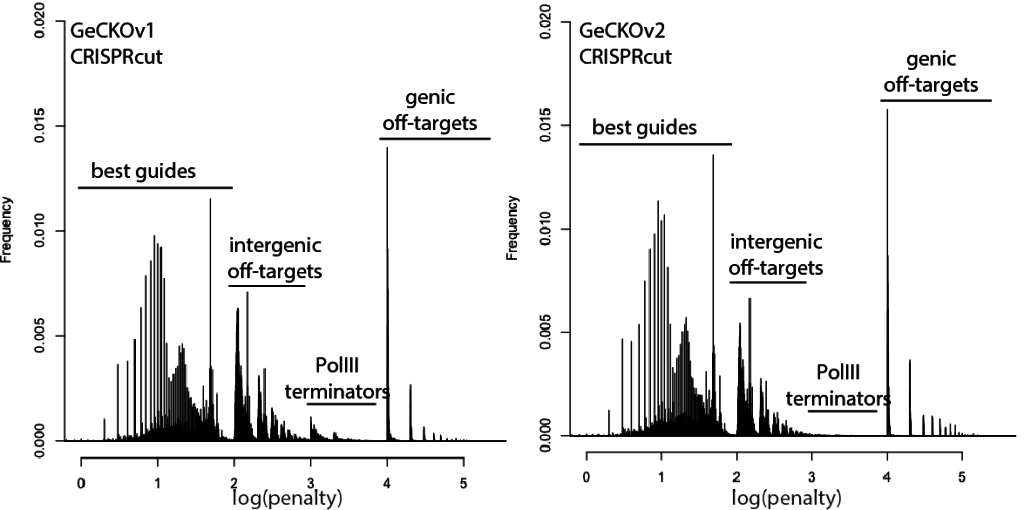
Is Cas9 specific?
I started writing a lengthy analysis about whether or not Cas9 is specific. It contained several in-depth analyses of many papers. There were arguments for and against. But right in the middle I...
I started writing a lengthy analysis about whether or not Cas9 is specific. It contained several in-depth analyses of many papers. There were arguments for and against. But right in the middle I realized that the details of the literature surrounding specificity don't really matter and I deleted the whole thing. Here's why...
Cas9's specificity might be pretty interesting to you if you're creating cell lines for research use. After all, you don't want to be reporting a phenotype that actually stems from some off-target knockout. But if you're thinking about a gene correction therapy, specificity will keep you up at night in a cold sweat.
And that distinction, together with the mindset of research vs therapeutics, is the key. Several papers have shown that Cas9 is both moderately specific and moderately permissive. There's all kinds of literature about seed regions and sgRNA bubbles, and so on. But right now, we can say that sgRNAs can be at least pretty good, and it's hard to tell when they'll be bad.
So for research purposes, it doesn't matter whether or not Cas9 is highly specific, because you should just choose two distinct guides and demonstrate that your phenotype is robust to choice of guides. The chance that two guides of different sequence will have the same off-target is very low. This is much like what's done with genome-wide CRISPRcut/i/a libraries, but I think it should also extend to CRISPR-made cell lines and probably even animal models.
And for therapeutic purposes, it doesn't really matter what the literature says. Even if all papers everywhere said that Cas9 was absolutely stringent, you'd still need to demonstrate specificity (or at least knowledge of benign off-targets) for your application of interest. Anyone who would use a genome-targeting reagent in humans without careful homework on that reagent (regardless of literature precedent) has no business making therapeutics.
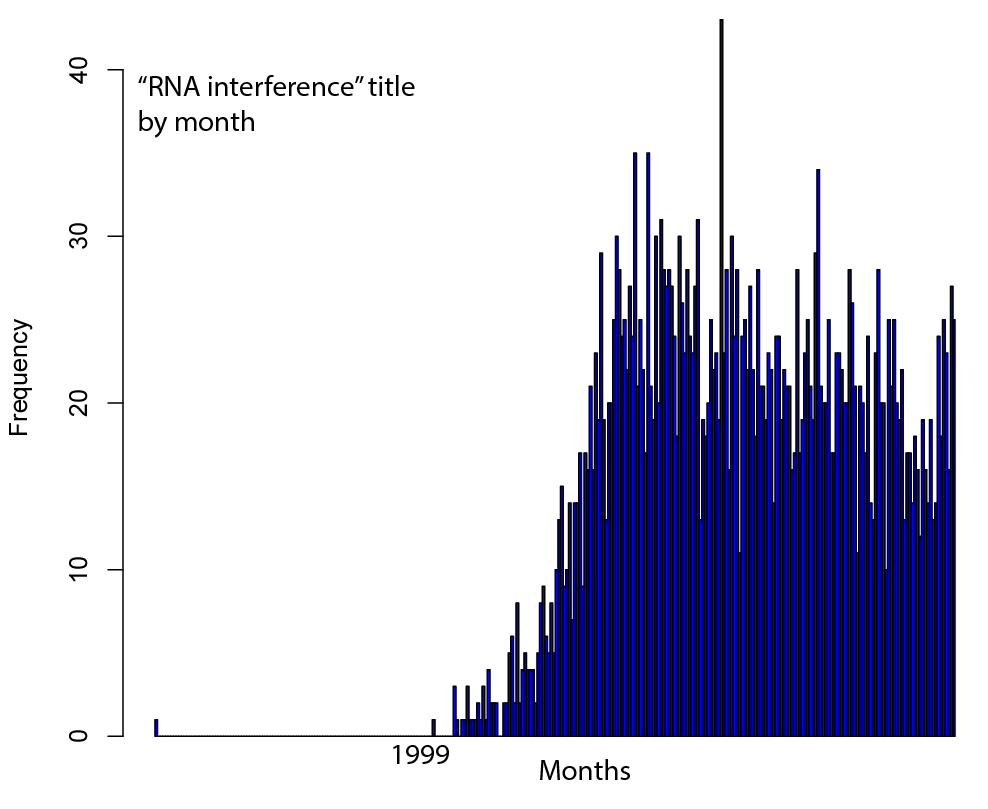
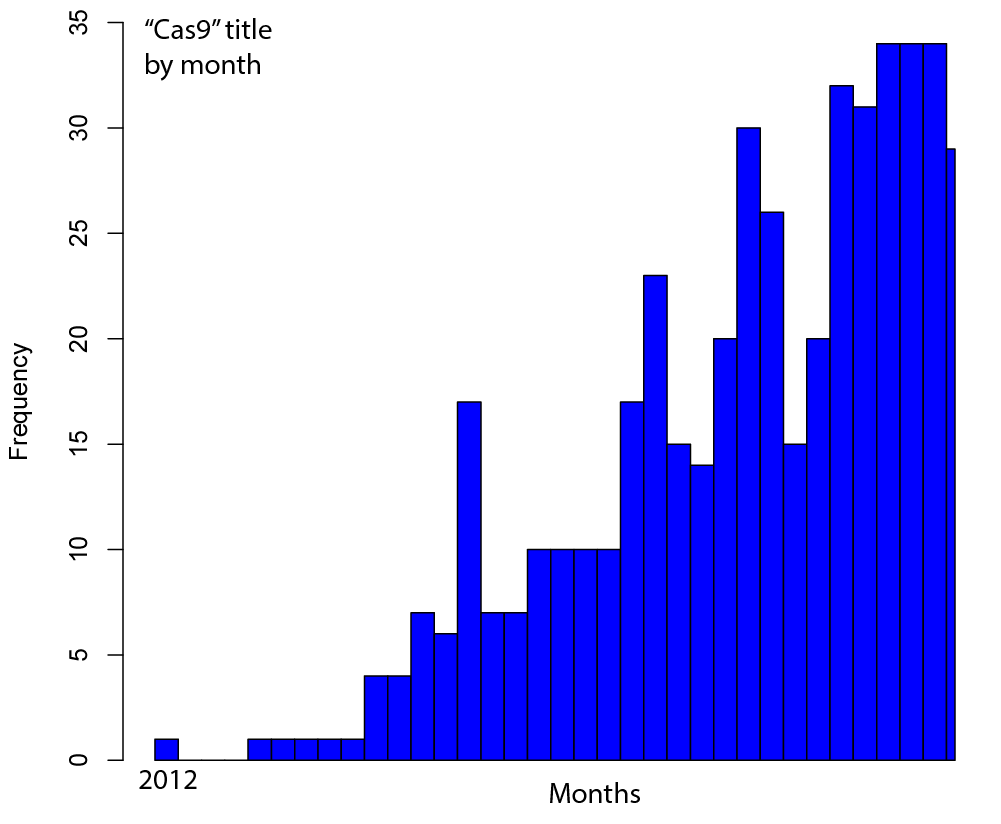
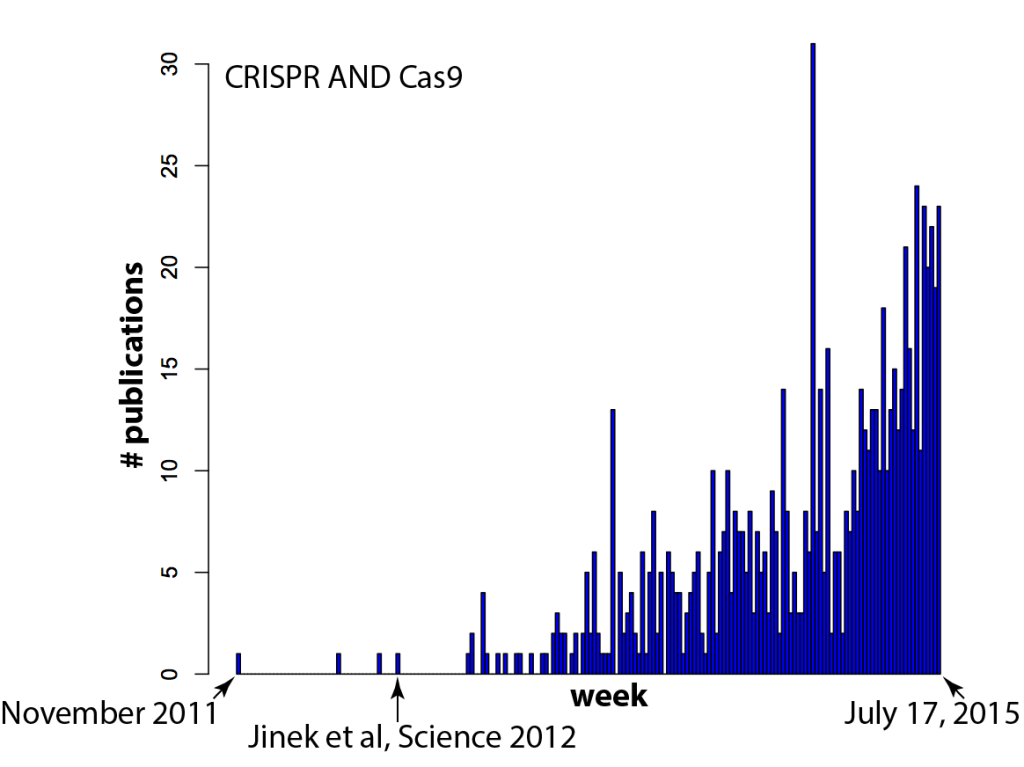
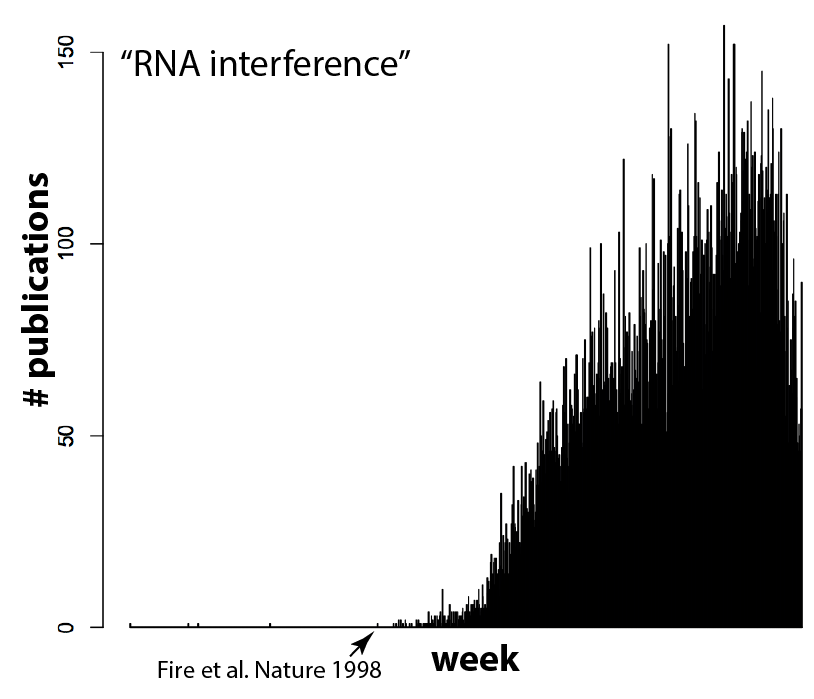


 Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...
Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...