This post is all about establishing safety for CRISPR gene editing cures for human disease. Note that I did not say this post is about gene editing off-targets. We’ll get there, but you might be surprised by what I have to say.
Contrary to what some might say or write, most of us gene editors do not have our heads in the sand when it comes to safety. From a pre-clinical, discovery research point of view, the safety of a given gene editing technology is relatively meaningless. There are many dirty small molecules out there that you’d never want to put in a person but are ridiculously useful to help unravel new biology. Given that pre-clinical researchers have the luxury of doing things like complementation/re-expression experiments and isolating multiple independent clones, let’s dissociate everyone’s personal research experience with CRISPR (all pre-clinical at this point) from questions of safety. Those experiences are useful and informative, but so far anecdotal and not necessarily tightly linked to the clinic.
Despite what we gene editors like to think, CRISPR safety is not a completely brand new world full of unexplored territory. While there are some important unanswered questions, there’s a lot of precedent. Not only are other gene editing technologies already in the clinic, but non-specific DNA damaging agents are actually effective chemotherapies (e.g. cisplatin, temozolomide, etoposide). In the latter case, the messiness of the DNA damage is the whole point of the therapy.
Here are a few thoughts about the safety of a theoretical CRISPR gene editing therapy, in mostly random order. I’ll preface this by saying that, while I have experience in the drug industry, I’m by no means an expert on clinical safety and defer to the real wizards.
Safety is about risk vs reward
Let’s start with a big point: as with any disease, the safety of a gene editing therapy is all about the indication. The safety profile of a treatment for a glioblastoma (very few good treatment options for a fatal disease with fast progression) will be very different than a treatment for eczema. And the safety tolerance of a glioblastoma treatment that increases progression-free survival by only two days is going to look different than one that increases overall survival by five years. So there won’t be One True Rule for gene editing safety, since most of the equation will be written by the disease rather than the therapy.
Gene editing safety is about functional genotoxicity
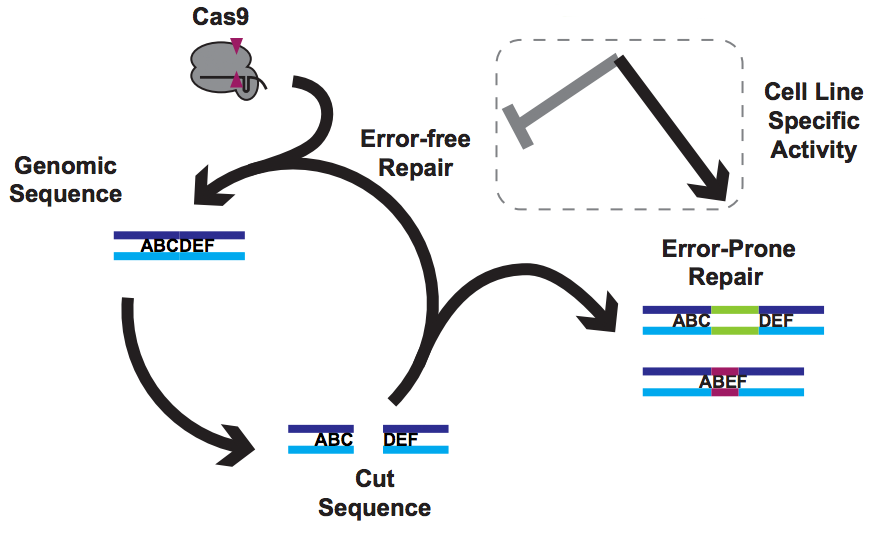
While most of the safety equation is about the disease, the treatment itself of course needs to be taken into account. At heart, gene editing reagents are DNA damaging agents, and so genotoxicity is a big concern. Does the intervention itself disrupt a tumor suppressor and lead to cancer? Does it break a key metabolic enzyme and lead to cell death? As mentioned above, there are plenty of DNA damaging agents that wreak havoc in the genome, but are tolerated because due to risk/reward and lack of a better alternative (I’m especially looking at you, temozolomide). The key to this point is function of what gets disrupted.
With CRISPR, we have the marked advantage that guide RNAs tend to hit certain places within the genome. We know how to design the on-target and are still figuring out how to predict and measure the off-target. But even with perfect methods for off-targets, we’d still need to do the functional test. Consider a “traditional” therapeutic (small molecule or biologic) – while an in vitro off-target panel based on biochemistry is valuable, it’s no substitute at all for normal-vs-tumor kill curves (as an example). And even those kill curves are no substitute for animal models. The long term, functional safety profile of a gene editing reagent is the key question, and with CRISPR I’d argue that we’re still too early in the game to know what to expect. The good news is that ZFNs so far seem pretty good, giving me a lot of hope for gene editing as a class.
Gene editing safety is NOT about lists of sequences
You’d think that determining an exhaustive list of off-target sequences would be a critical part of any CRISPR safety profile. But in the example above, contrasting in vitro biochemical assays with organismal models, I consider lists of off-targets to be equivalent to the biochemical assay. I’m going to be deliberately controversial for a moment and posit that, for a therapeutic candidate, you shouldn’t put much weight on its list of off-target sites. As stated above, you should instead care about what those off-targets are doing, and for that you might not even need to know where the off-targets are located.
When choosing candidate therapeutics in a pre-clinical mode, lists of off-target sequences can be very useful in order to prioritize reagents. If one guide RNA hits two off-target sites and another hits two hundred, you’d probably choose the former rather than the latter. But what if one the two off-targets is p53? What if the two-hundred are all intergenic? Given the fitness advantage to oncogenic mutations, the math involved in using sequencing (even capture-based technologies) to detect very rare off-target sites is daunting. Being able to detect a 1 in a million sequence-based event sounds incredible, but what if you need to edit as many as 20 million cells for a bone marrow transplant? That’s twenty cells you might be turning cancerous but never even know it. Now we come right back around to function – you should care much more about the functional effect of your gene edit rather than a list of sequences. That list of sequences is nice for orders-of-magnitude and useful to choose candidate reagents, but it’s no substitute for function.
Is gene editing safety about immunogenicity?
There are two big questions around gene editing immunogenicity: the immunogenicity of the reagent itself, and on-target immunogenicity if the edit introduces a sequence that’s novel to the patient. What happens when the reagent itself induces a long-term immune response? For therapies that require repeat dosing, this can kill a program (hence a huge amount of work put into humanizing antibodies). A therapy that causes someone to get very sick on the second dose is not much good, nor is it useful if antibodies raised to the therapy end up blocking the treatment. But what about in situ gene editing?
Most of in situ gene editing reagents are synthetic or bacterial and so one might raise antibodies against them, but the therapy itself is (ideally), one-shot-to-cure. In that case, as long as there’s not a strong naive immune response, maybe it doesn’t matter if you develop antibodies to the editing reagent? There are few answers here for CRISPR, and most work with ZFNs has been with ex vivo edits, where the immune system isn’t exposed to the editing reagent. Time will tell if this is a problem, and animal models will be key. Even more subtle, what happens when a gene edit causes re-expression of a “normal” protein that a patient has never before expressed (e.g. editing the sickle codon to turn mutant hemoglobin into wild type hemoglobin)?
The potential for a new immune response against a new “self” protein is probably related to the extent of the change – a single amino acid change (e.g. sickle cell) is probably less likely to cause problems than introducing a transgene (e.g. Sangamo’s work inserting enzymes into the albumin locus for lysosomal storage disorders and hemophilia). But once again, I’ve heard a lot of questions and worry about this problem but very few answers. In vivo experiments are desperately needed, and the closer to a human immune system the better.
Moving forward based on the predictive data
As you’ve probably gathered by now, I have a healthy respect for functional characterization when it comes to safety. That’s why it’s absolutely critical that we keep moving forward and not let theoretical worries about arbitrary numbers of off-targets stifle innovation without data. These are tools that could some day help patients in desperate need and with few other options, so let the truly predictive functional data rule the day.
X Close
 Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...
Welcome to Lena Kobel, who joins the lab as a Cell Line Engineer. Lena has a long history in genome engineering, with previous experience in Martin Jinek’s...
 We also wanted our approach to be as general as possible – to develop something that works for SCD but is accessible to clinical researchers everywhere. That would fit the democratization theme of Cas9. Both the nuclease targeting reagent and the approach to allele replacement would be fast, cheap, and easy for everyone to use, so that everyone could ask questions about their own system in HSCs and maybe even develop gene editing cures for the particular disease in which they’re working.
We also wanted our approach to be as general as possible – to develop something that works for SCD but is accessible to clinical researchers everywhere. That would fit the democratization theme of Cas9. Both the nuclease targeting reagent and the approach to allele replacement would be fast, cheap, and easy for everyone to use, so that everyone could ask questions about their own system in HSCs and maybe even develop gene editing cures for the particular disease in which they’re working.