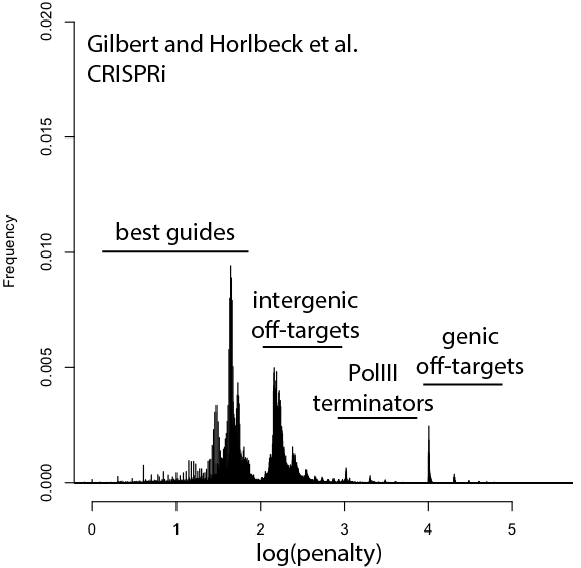
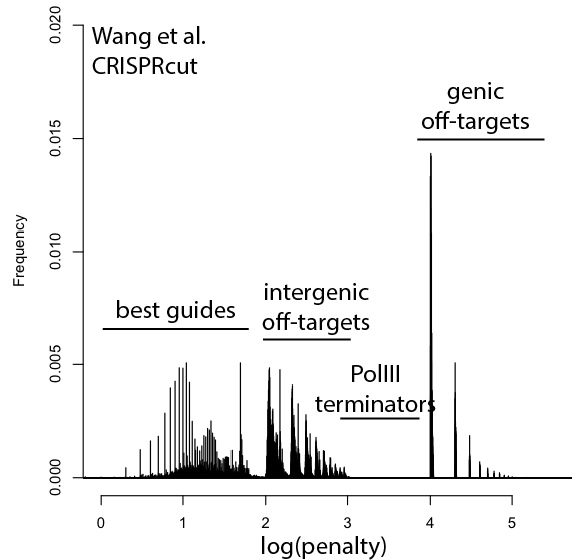
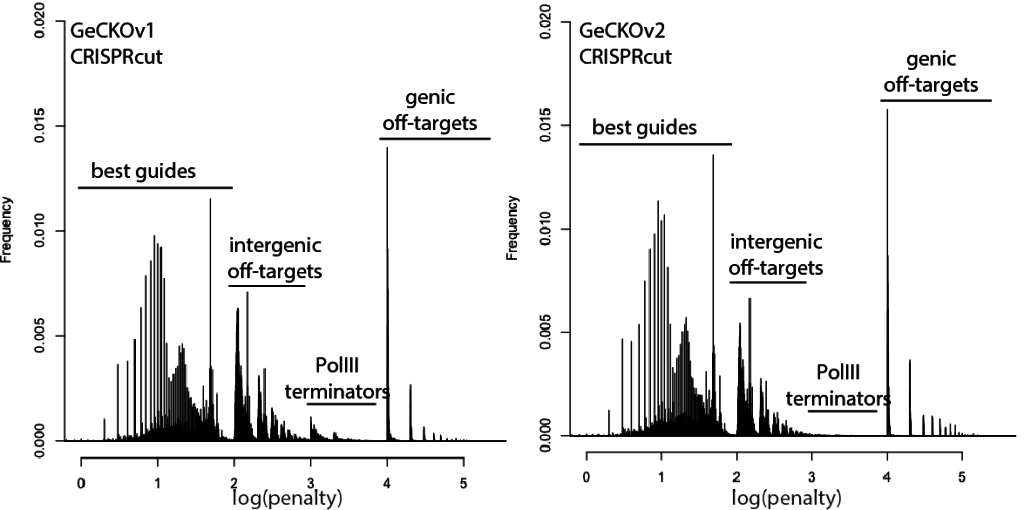
A wish list for science
File this one under “crotchety grumblings.” I started this post as a wish list for the future of CRISPR/Cas9, but came to the realization that ...
File this one under “crotchety grumblings.” I started this post as a wish list for the future of CRISPR/Cas9, but came to the realization that it could apply to just about any sexy field. So instead, this is a wish list for biology in general. These points are related to the dichotomy between the business of science and the pursuit of knowledge. I’m sure some of these points have come up in previous posts, but I want to get this off my chest in one place. I understand that the business of science has very logically led us to a point where the items below are natural and common, but it doesn’t make them right. I’m also sure that most of these have been discussed in one form or another elsewhere, but I’m adding my $0.02. Consider this a reminder for students and postdocs who might be reading the blog.
At heart, all of the below come down to one thing. As scientists, we are trying to discover new things about the world. One assumption to most of our work is that there is an external, objective truth for us to observe. We see that truth through a glass darkly, and hence we attempt to put the external world in a form that can be understood from human context. We can do no else. To purposefully distort that truth does real harm and sets everyone back, but has unfortunately become de rigueur in some contexts.
- Rickety Tricks. Doing tech dev or poking in to biology by taking advantage of overly-specific proof of concept just creates frustration and eventually casts a black cloud over whole fields. Science is typically bleeding edge, and it can be very difficult to know the boundaries of an approach while it’s being developed. But that does’t justify purposefully take advantage of “tricks” to look successful while simultaneously knowing that there’s no clear path anywhere other than the one-off proof of concept experiment. This cynical approach might inflate one’s own CV, but generates mistrust for whole fields and poisons the potential for real world application.
- Reproducibility. Related to the above, there have been several articles about the reproducibility problem in pre-clinical science. I won’t bother to link to all of them, since by this point the problem is well-recognized and I could only accidentally miss some attributions. The general problem is that perverse incentives for publication mean that many pieces of research are just plain wrong. This isn’t necessarily outright fraud, but it can stem from squinting at the data in just the right way and hoping, because positive results are good but there’s no Journal of Negative Results. I saw much less of this when I was in industry, perhaps because the motivations are much different. If one is just aiming to get a paper out the door, then one might not particularly care if a discovery is Right or Reproducible (though one should!). But in industry, that paper (or internal discovery) is just the start for what might be a very expensive and long-running drug discovery project, and there are many incentives for robustness and reproducibility in that case.
- Over Hyping. Be honest those who consume your science (both primary articles and press releases + interviews) and yourself. As we approach an age of open science, it’s becoming easier for non-scientists to access research articles and learn about cutting-edge developments. This is a very good thing, since non-scientists are frequently very motivated, for example by family members suffering from a disease. These people should have access to all the information they might want. But coming from outside the business of science, they might not understand the hype machine. Promising the world based on an incremental advance might increase one’s chances for getting into Nature, but has very real consequences by potentially giving people false hope. I feel very strongly here, since I’ve personally answered emails and telephone calls from people who saw certain overhyped papers and called me to ask about cures for their sick child. It’s heart-breaking. And if compassion for fellow humans doesn’t motivate, consider that the government (including people who set budgets!) have little tolerance for science that promises then doesn’t deliver.
It’s a beautifully sunny day on the weekend, so I’ll stop here before I dip into too much of a funk. But I’ll end by saying that these are things we can fix. There is no Science with a capital S. We are scientists who are building science as we go along! We are a global community – we write our own papers, referee our own papers, write our own blog posts, form our own societies, and organize our own conferences. It’s all up to us.